Tính chất điện tử của Hexagonal Chromium Nitride
Trong nghiên cứu này, phương pháp gần đúng liên kết mạnh (TB) được sử dụng để xây dựng mô hình Hamiltonian tính toán đặc trưng điện tử của hexagonal chromium nitride (h-CrN) cho cấu trúc phẳng và nhấp nhô.
Ngày nay, vật liệu mỏng hai chiều như graphene và tựa graphene đang nhận được sự quan tâm đặc biệt của các nhà nghiên cứu lý thuyết và thực nghiệm, bởi các đặc tính vật lý thú vị của nhóm vật liệu này và tiềm năng ứng dụng mạnh mẽ trong công nghiệp bán dẫn và transistor. Tuy nhiên, đối với các ứng dụng công nghiệp liên quan đến sự phân cực của spin thì nhóm các vật liệu hai chiều có cấu trúc là một lớp nguyên tử này lại không phải là đối tượng hàng đầu nhận được sự quan tâm. Bởi đây là nhóm vật liệu không có hoặc có từ tính rất yếu. Trong khi đó, các vật liệu ở nhóm kim loại chuyển tiếp (transition metal nitrides-TMNs) hoàn toàn có thể khắc phục được nhược điểm của nhóm vật liệu hai chiều này. Ngoài những tính chất vật lý giống với nhóm vật liệu hai chiều, các vật liệu thuộc TMNs còn có khả năng hình thành các trạng thái spin bề mặt (do cấu trúc vật liệu tồn tại electron ở phân lớp d), rất thuận lợi cho việc ứng dụng vào các thiết bị spintronic.
Trong số đó, với cấu trúc tinh thể được coi là một thành viên của hệ TMNs trong khi cấu trúc theo hướng (111) lại là một màng mỏng như một lớp màng tổ ong của nhóm vật liệu 2D (dạng hexagonal), Chromium Nitride (CrN) có thể nói là sự kết hợp hoàn hảo của những tính chất đặc biệt của 2 hệ vật liệu lớn này, hứa hẹn nhiều ứng dụng cho ngành công nghiệp điện tử. Tuy nhiên, số lượng nghiên cứu lý thuyết về vật liệu này còn khá khiêm tốn, đa phần tập trung ở các phương pháp thực nghiệm. Do đó, nhu cầu về việc phát triển nghiên cứu lý thuyết của vật liệu này là điều thực sự cần thiết để có thể dự đoán những tính chất đặc biệt của vật liệu dưới các tác nhân kích thích bên ngoài cũng như lựa chọn những khả năng để đưa chúng vào ứng dụng.
Trong nghiên cứu này, bằng việc sử dụng phương pháp gần đúng liên kết mạnh để xây dựng mô hình tính toán cho các tương tác lân cận gần nhất của các orbital nguyên tử, bài toán cấu trúc vùng của vật liệu CrN được khảo sát cho cả hai trạng thái: trạng thái phẳng và trạng thái nhấp nhô; đồng thời, so sánh với kết quả thu được từ các nghiên cứu trước để đề xuất bộ tham số cấu trúc phù hợp, hướng đến việc sử dụng trong các tính toán tiếp theo, liên quan đến các tương tác spin của vật liệu. Vì thế, đây có thể là nghiên cứu mở đường cho chuỗi những nghiên cứu sau này về nhóm các vật liệu đang có giá trị ứng dụng vào cuộc cách mạng công nghiệp như trong thời điểm hiện nay.
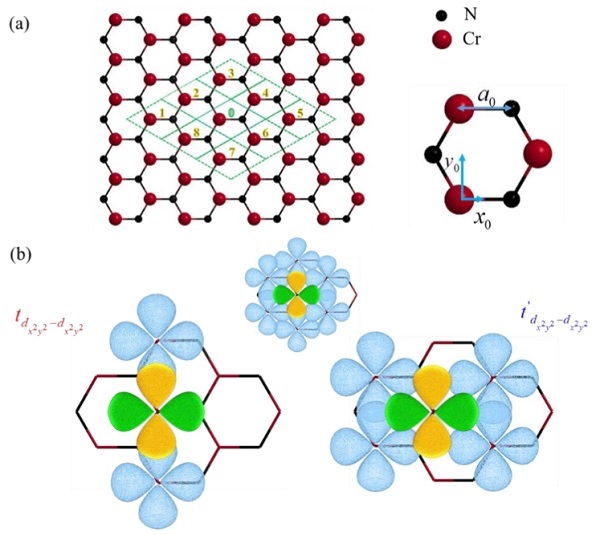
Mô hình tính toán h-CrN dựa trên phương pháp gần đúng liên kết mạnh, (b) Tương tác giữa các orbital của nguyên tử Cr
Trong bài báo này, mô hình tính toán được xây dựng và chỉ ra đóng góp của các orbital trong việc hình thành các tính chất điện tử đặc biệt của h-CrN, dựa trên phương pháp gần đúng liên kết mạnh và phương pháp luận hàm Green. Đồng thời, bộ tham số cấu trúc về năng lượng tương tác giữa các orbital của nguyên tử Cr và N cho mô hình phẳng và nhấp nhô cũng được trình bày. Kết quả nghiên cứu cho thấy rằng ở trạng thái nhấp nhô, cùng với sự đóng góp của các orbital nguyên tử N gần nhau nhất, trạng thái dẫn của vật liệu được kích thích thay đổi nhiều hơn so với trạng thái phẳng ban đầu. Đồng thời, tham số ảnh hưởng liên quan đến tính chất spin của vật liệu cũng được khảo sát. Đây là những kết quả bước đầu trong việc khảo sát tính chất điện tử của vật liệu CrN bằng phương pháp gần đúng liên kết mạnh, làm tiền đề để nghiên cứu những tính chất khác nhau của vật liệu dưới tác dụng của kích thích bên ngoài như điện trường, hướng tới ứng dụng trong tương lai.
Tạp chí Khoa học Trường Đại học Cần Thơ, Tập 57, Số 6A (2021)